
Զարգացման հապաղում, եղջերաթաղանթի պղտորում, հիպոմիելինացնող լեյկոէնցեֆալոպաթիա. հերքել IV տիպի մուկոլիպիդոզը
Մանե Թադևոսյան 1, Բիայնա Սուխուդյան 1,2, Դավիթ Բաբիկյան 3, Օյգեն Բոլտսհաուզեր 4
1 Մանկական նյարդաբանության բաժանմունք, Արաբկիր բժշկական համալիր, Երևան, Հայաստան
2 Նյարդաբանության և նյարդավիրաբուժության ամբիոն, Ակադեմիկոս Ս.Ավդալբեկյանի անվան առողջապահության ազգային ինստիտուտ, Երևան, Հայաստան
3 Բժշկական գենետիկայի և առողջության առաջնային պահպանման կենտրոն, Երևան, Հայաստան
4 Նյարդաբանւթյան բաժանմունք, Համալսարանական մանկական հիվանդանոց, Ցյուրիխ, Շվեյցարիա
ԱՄՓՈՓԱԳԻՐ
IV տիպի մուկոլիպիդոզը (MLIV) ծայրահեղ հազվադեպ հանդիպող, աուտոսոմ ռեցեսիվ ժառանգմամբ, լիզոսոմային հիվանդություն է, որը բնութագրվում է բնորոշ նյարդաբանական (վաղ սկսվող զարգացման հապաղում, սպաստիկություն), ակնային (եղջերաթաղանթի պղտորում, ռետինոպաթիա) և բնորոշ ՄՌՇ նշաններով (հիպոմիելինացնող լեյկոէնցեֆալոպաթիա, բարակ բրտամարմին, ուղեղիկի ատրոֆիա): Թեև MLIV-ը հիմնականում նկարագրվում է աշքենազի հրեական համայնքում, այն համաէթնիկական հիվանդություն է։ Հեղինակները ներկայացնում են ազգությամբ հայ երկու եղբայրների, որոնք ունեն MLIV-ին բնորոշ գլոբալ զարգացման հապաղում, ակնային շեղումներ և ՄՌՇ պատկեր։ Ամբողջական էքզոմի սեքվենավորումը հայատնաբերել է MCOLN1 գենի երկու ախտածին տարբերակ: Այսպիսով, հաստատվում է, որ կլինիկական և ՄՌՇ պատկերների նման համախումբը հիմք է հանդիսանում MLIV-ի ախտորոշման համար, և առաջարկվում է նպատակային ախտորոշիչ հետազոտություններ կատարել անհայտ ծագման նյարդային զարգացման խանգարումների դեպքում:
Հիմնաբառեր. IV տիպի մուկոլիպիդոզ, MCOLN1, եղջերաթաղանթի պղտորություն, հիպոմիելինացում, մանկական ուղեղային կաթված
DOI: 10.54235/27382737-2023.v3.1-52
INTRODUCTION
Mucolipidoses (MLs) are classified as lysosomal storage diseases (LSDs) because of their involvement in increased storage materials in the lysosomes [1]. The group includes four diseases: ML I (sialidosis), ML II (inclusion-cell, or I-cell, disease), ML III (pseudo-Hurler polydystrophy), and ML IV, grouped together due to similar clinical manifestations and storage pattern, yet arising from mutations in distinct genes [2]. Mucolipidosis type IV (MLIV) (OMIM 252650) is a rare autosomal recessive LSD resulting from loss-of function mutations in the MCOLN1 gene [3,4].
MLIV manifests with severely impaired neurodevelopment, and slowly progressive, gradual neurological, and ophthalmological abnormalities, including corneal opacities, retinal degeneration, and strabismus [5,6].
In addition to ophthalmological findings, brain magnetic resonance imaging (MRI) plays an important role in diagnosis of MLIV. MRI studies reveal hypomyelination and, consequently, thin corpus callosum, decreased signal intensity in the basal ganglia and thalami, and cerebellar atrophy [3,7].
The clinical diagnosis of MLIV can be challenging as it initially presents with impaired neurodevelopment in the absence of progressive features till later (towards end of first and in second decade) in life. Patients with MLIV can be misdiagnosed as having cerebral palsy, non-compressive myelopathy, spastic paraplegia, and other disorders [7].
Apart from drawing attention to a rare genetic disorder the aim of this case report is to highlight that clinical reasoning may allow targeted additional (i.e. genetic) investigations, and to recommend that unsolved developmental disorders (as “atypical” cerebral palsy) need a careful diagnostic work-up.
CASE STUDY
Patient 1
The child was born from the third pregnancy and second delivery. There was no consanguinity. The second pregnancy ended in a spontaneous abortion. The male child was delivered at full term by vaginal delivery with appropriate birth weight (2450 g – 3 percentile), height (48 cm), head circumference (33 cm), and normal Apgar scores. Global neurodevelopmental delay was noticed by parents during infancy. He had sucking, chewing and swallowing problems. Independent sitting was achieved at the age of 2 years. After that, a slow regression of the achieved developmental skills was observed.
Neurological examination was first done at the age of 7 years. His head circumference was 51.5 cm (10-25p). The child had no words. Neurological examination showed alternating convergent strabismus. Eye tracking movements were normal with marked intention tremor. Hypotrophy of thenar and hypothenar muscles was visible. Hands were in permanent stereotyped movements. Muscle strength was normal. There was a right foot equinovarus deformity with fixed contracture. Muscle tone and deep tendon reflexes were increased. Physical examination revealed dry skin.
Complete blood count revealed iron-deficiency anemia with a hemoglobin level of 109 g/L. Ophthalmological examination revealed corneal clouding and partially pale optic discs. Abdominal ultrasound was normal.
MRI was performed at the age of 7 years (Figures 1 and 2). Myelination was not age-appropriate but compatible with hypomyelinating leukoencephalopathy; lack of myelin in the temporal lobes and internal capsule, many cerebral gyri were not myelinated, reduced myelination in the white matter bulk of the frontal lobes. As a result of hypomyelination, the corpus callosum was very thin, but all elements (genu, splenium) were present. Abundant perivascular Virchow-Robin spaces (VRS) were present throughout the centrum semiovale, some were cystic. There was cerebellar atrophy.
Patient 2
The boy, the brother of Patient 1, was the third child in the family, born from the fourth pregnancy. The perinatal history was uneventful. According to the mother, the development of this child was normal until 11 months. He could sit independently, mental development was also normal, he even had some words, later regression of both motor and mental skills was observed. No seizures were reported.
At the age of 3 years he was found to have severe iron-deficiency anemia. At the age of 6 years the neurological examination revealed horizontal nystagmus on abduction, lack of independent sitting, inadequate laughing or crying. Increased muscle tone and deep tendon reflexes with enlarged reflexogenic zones were observed. Physical examination revealed dry skin. Abdominal ultrasound was normal.
MRI was performed at the age of 3 years. The findings were similar to those in Patient 1. Remarkably, cerebellar atrophy was already evident at this age.
Genetic evaluation
MLIV was suspected based on the clinical constellation with early-onset developmental deviation, spasticity, corneal clouding, and neuroimaging evidence of a hypomyelinating leukoencephalopathy with cerebellar atrophy. Due to financial constraint, genetic testing was done in Patient 1 only. Whole-Exome Sequencing of Patient 1 revealed compound heterozygous pathogenic variants in MCOLN1: (c. 514C>T(p. Arg172) and (c.985-2A>G(p.?)). According to the 2015 American College of Medical Genetics and Genomics (ACMG 2015) criteria, the MCOLN1(c. 514C>T (p. Arg172) variant is considered a pathogenic variant. In ClinVar database too, it is listed as a pathogenic variant (ID: 208030). This variant was reported before in patients with MLIV.
According to ACMG 2015 criteria, the MCOLN1 (c.985-2A>G(p.?) variant is considered pathogenic. It is not registered in either GnomAD or ClinVar databases. This variant has not been reported before.
DISCUSSION
Two siblings of Armenian descent affected by Mucolipidosis type IV are reported.
To the best of our knowledge this is the first report of MLIV in Armenia. The constellation of neurological (early-onset developmental delay, spasticity), ocular (corneal opacity, retinopathy), and characteristic MRI findings (hypomyelinating leukoencephalopathy, thin corpus callosum, cerebellar atrophy) is very characteristic for MLIV [8].
Clinical considerations
Often the clinical diagnosis of MLIV can be a challenge. There are no pathognomonic clinical signs for MLIV when taken in isolation. Considering the initially static and then the slow progressive course, the patients can be misdiagnosed as having cerebral palsy, non-compressive myelopathy, spastic paraplegia, and other disorders [3]. Delayed motor development with hypotonia is frequently the earliest neurologic feature of MLIV [7]. Later in course of the disease, slowly progressive neurological abnormalities are observed: in particular spasticity of limbs. Individuals with typical MLIV have superficial corneal opacity that is bilateral, symmetric, and most visible in the central cornea [7]. As corneal clouding is the first noticeable sign, it is often an important diagnostic clue in MLIV. Corneal clouding can be found in other LSDs, in particular in some types of mucopolysaccharodosis and in GM1 gangliosidosis, but the overall context allows distinction from MLIV. Affected individuals do not have hepatosplenomegaly or specific skeletal abnormalities [7]. However, a number of storage disorders (as NCL, M. Fabry) share this lack of organomegaly.
Neuroimaging considerations
Besides neurological and ocular abnormalities, characteristic brain MRI findings are important clues to the diagnoses of MLIV. The finding of white matter abnormalities and a thin dysplastic corpus callosum could suggest other inherited hypomyelinating leukodystrophies [7].
Dilated VRSs are a common finding in many disorders, syndromes and even healthy controls. As an example they are characteristic for mucopolysaccharidosis, where, typically, the VRSs are dilated by accumulated glycosaminoglycans, which results in a cribriform appearance of the white matter, corpus callosum, and basal ganglia on T1-weighted images [9]. One of the MRI characteristics may be the decreased T2-signal intensity of the basal ganglia and thalami in MLIV patients, which was not observed in the presented two children [10]. This reduced signal is due to iron deposited in the brain in the form of ferritin [8,11].
Atrophy of the cerebellum and cerebrum is one of the hallmarks of MLIV and has been previously observed in older patients, which may reflect the disease progression [8]. However, in the Patient 2, cerebellar atrophy was seen already at the age of 3 years.
Other diagnostic investigations
Other diagnostic tests may be helpful for further diagnosis. Individuals with MLIV present with iron-deficiency anemia, which can be easily confirmed. Normal gastric acid production is affected in patients with MLIV. This produces a constitutive achlorhydria, which leads to iron-deficiency anemia and high serum gastrin level via the feedback pathway [12,13]. Blood gastrin measurement or skin biopsy has often been used to confirm the diagnosis of MLIV in the past [12]. Characteristic lysosomal storage bodies can be visualized by electron microscopy in skin or conjunctiva biopsy [13].
The diagnosis is finally confirmed by molecular genetic testing.
Therapeutic considerations
Treatments for some LSDs have included enzyme replacement therapy, substrate reduction therapy, and gene therapy [14]. Unfortunately, there is no specific treatment for MLIV. Treatment of MLIV is symptomatic and supportive. Physical therapy with special focus on spasticity and ataxia can improve motor function. Iron supplementation is utilized for those with anemia.
CONCLUSION
Mucolipidosis type IV is a rare autosomal recessive lysosomal storage disorder that should be considered in case of unexplained neurodevelopmental disorders and which may be suspected based on specific clinical and MRI features. The diagnosis of this disease is crucial for the family planning. For at-risk families with known genotypes, prenatal or preimplantation genetic testing may be offered.
ACKNOWLEDGMENTS
The authors would like to thank the family for participating and supporting this study.
REFERENCES
- Khan SA, Tomatsu SC. Mucolipidoses overview: past, present, and future. Int J Mol Sci. 2020;21(18):6812.
- Boudewyn LC, Walkley SU. Current concepts in the neuropathogenesis of mucolipidosis type IV. J Neurochem. 2019;148(5):669-689.
- Wakabayashi K, Gustafson AM, Sidransky E, Goldin E. Mucolipidosis type IV: An update. Mol GenetMetab. 2011;104(3):206-213.
- Jezela-Stanek A, Ciara E, Stepien KM. Neuropathophysiology, genetic profile, and clinical manifestation of mucolipidosis IV – A review and case Series. Int J Mol Sci. 2020;21(12):4564.
- Misko A, Wood L, Kiselyov K, Slaugenhaupt S, Grishchuk Y. Progress in elucidating pathophysiology of mucolipidosis IV. Neurosci Lett. 2021;755:135944.
- Sun M. Mucolipidosis type IV is caused by mutations in a gene encoding a novel transient receptor potential channel. Hum Mol Genet. 2000;9(17):2471-2478.
- Misko A, Grishchuk Y, Goldin E, Schiffmann R. Mucolipidosis IV. In: Adam MP, Everman DB, Mirzaa GM, et al., eds. GeneReviews®. University of Washington, Seattle; 1993.
- Frei KP, Patronas NJ, Crutchfield KE, Altarescu G, Schiffmann R. Mucolipidosis type IV: characteristic MRI findings. Neurology. 1998;51(2):565-569.
- Kwee RM, Kwee TC. Virchow-Robin Spaces at MR Imaging. RadioGraphics. 2007;27(4):1071-1086.
- D’Arco F, Hanagandi P, Ganau M, Krishnan P, Taranath A. Neuroimaging findings in lysosomal disorders: 2018 update.
Top Magn Reson Imaging. 2018;27(4):259-274. - Gelman BB. Iron in CNS Disease: J Neuropathol Exp Neurol. 1995;54(4):477-486.
- Al-Alawi B, Harikrishna B, Al-Thihli K, et al. Mucolipidosis type IV in Omani families with a novel MCOLN1 mutation: search for evidence of founder effect. Genes. 2022;13(2):248.
- Geer JS, Skinner SA, Goldin E, Holden KR. Mucolipidosis type IV: A subtle pediatric neurodegenerative disorder. Pediatr Neurol. 2010;42(3):223-226.
- Hoffmann B, Mayatepek E. Neurological manifestations in lysosomal storage disorders – from pathology to first therapeutic possibilities. Neuropediatrics. 2005;36(5):285-289.
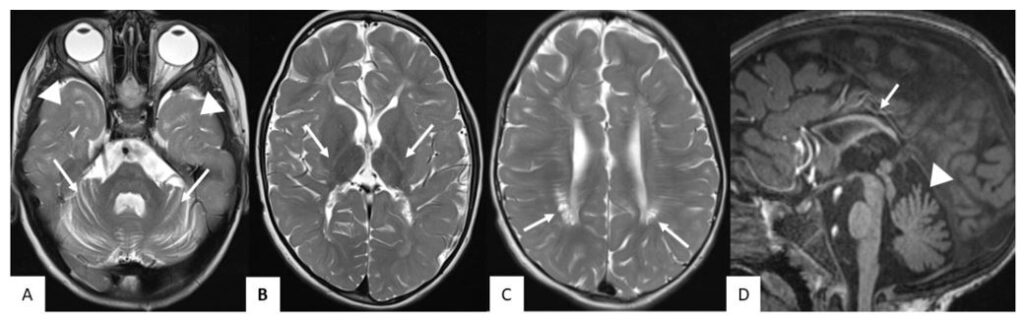
Figure 1. MRI of Patient 1 at the age of 7 years. Axial T2 images (A, B, C) show hypomyelination and no myelin in the temporal lobes (arrowheads in A) and the internal capsule (arrows in B). Abundant perivascular Virchow-Robin spaces in centrum semiovale were observed, some of them were cystic (arrows in C). Sagittal T1 image (D) shows thin corpus callosum (arrow in D) and cerebellar atrophy (arrowhead in D, also see arrows in A).
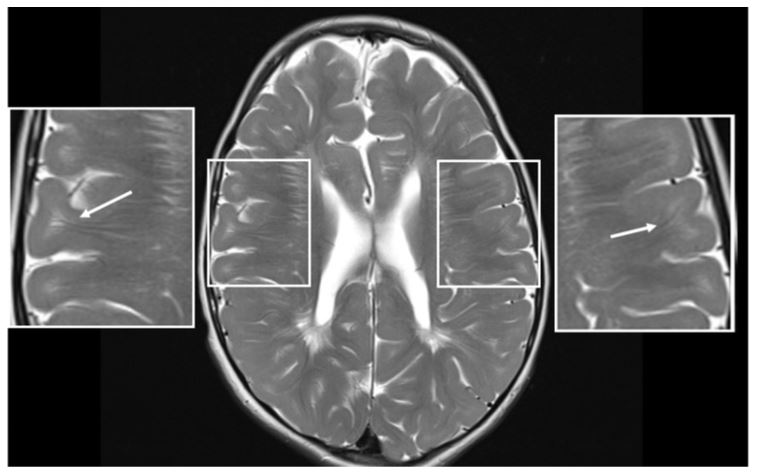
Figure 2. MRI of Patient 1 at the age of 7 years. Axial T2 image shows hypomyelination; many cerebral gyri are not myelinated (arrows).