

CASE DESCRIPTION
The patient is a 28-year-old otherwise healthy male. Four years prior to his admission, he noticed involuntary posture in his left hand, worsening of gait and speech, aggressive behavior, and irritability. On the first examination, he had inappropriate laughing, dysarthria, drooling of saliva, postural and kinetic tremor, more prominent on the left side, head and trunk tremor, dystonic postures in hands, bradykinesia, slow shuffling gait, and postural instability.
Due to the lack of high-resolution MRI, 0.35T MRI was performed – it did not reveal any lesions. Liver ultrasound was without any obvious abnormalities. The patient’s routine blood examination and aminotransferases were within normal limits. Plasma copper level was low – 2.0 μmol/L (lower normal limit – 13.4 μmol/L), and 24-hour urine copper was high – 1.8 μmol/24h (N=0.3-0.8 μmol/24h). The patient had Kayser–Fleischer rings (KFR) that were visible without additional ophthalmologic examinations (Fig. 1A). Unfortunately, the ceruloplasmin test was not available at that moment. The patient reported no family history of the neurological or hepatic disorder.
Diagnosis of Wilson’s disease (WD) was established, and the patient started penicillamine up to 750 mg per day divided into three doses with excellent control of motor function and partial improvement of behavioral symptoms. Now the patient is on penicillamine 250 mg with a stable effect. Currently, with effective treatment his KFRs substantially faded off (Fig. 1B). It is planned to switch to zinc acetate for maintenance treatment.
DISCUSSION AND BRIEF REVIEW
Wilson’s disease, also called hepatolenticular degeneration, is an inherited, autosomal recessive metabolic disorder caused by monogenic mutations in the ATP7B gene that results in the loss of function of copper transporter ATPase, impaired excretion of copper into the bile and subsequent accumulation of copper in the liver, and later in the brain and other organs. It is named after British neurologist Samuel Alexander Kinnier Wilson who described the association of neurological extrapyramidal symptoms with the liver disorder in his 1912 monograph dedicated to progressive hepatolenticular degeneration.
The prevalence of WD ranges between 1:30,000 and 1:40,000 [1,2] and can vary significantly across the world. Carrier frequency is from 1:90 to 1:100 and there is a 25% risk of inheritance of two mutations. The male to female ratio is 1.28 [3].
There is growing evidence that WD is much more prevalent than it was previously suggested. Large screening studies based on the ceruloplasmin plasma concentrations in Eastern Asia suggest a higher prevalence of WD – 1/1,500-1/3,000 [2,4]. A study on the genetic prevalence of WD conducted in UK showed that 1 in 40 is a heterozygote mutation carrier and the probable prevalence of WD can be as high as 1/7,000 [5]. This large gap between high genetic prevalence and a low number of diagnosed patients with WD can be explained by low penetrance of mutations and misdiagnosis still frequently encountered.
PATHOGENESIS AND COPPER METABOLISM
Alimentary copper is absorbed in a small intestine and transported through the cellular membrane into the enterocytes. Transport from enterocytes to bloodstream is performed by transmembrane protein APT7А. Copper is then transported to the liver through the portal vein, taken by hepatocytes where APT7B protein (APT7A is absent in hepatocytes) binds it to ceruloplasmin and transfers it into the blood. Excess copper is removed from hepatocytes into bile also by АТР7B. It is obvious how the deficit of ATP7B protein leads to copper accumulation in the liver. Ceruloplasmin is a protein that transports 95% of total copper. It is unstable and quickly degrades if not bound to copper and this is an explanation why low ceruloplasmin levels are detected in plasma in patients with WD. When copper intracellular concentration in hepatocytes exceeds the level of binding proteins, it damages hepatocytes. Free copper releases from the liver and targets various organs, such as the brain, eyes, red blood cells, and kidneys [6].
Copper in the brain acts through copper-dependent enzymes and normally participates in various functions, such as antioxidant defense, neurotransmitter metabolism and others. However, excess copper promotes oxidative stress and brain tissue damage. Copper is transported into the brain via endothelial cells of the blood-brain barrier and then released into the brain parenchyma by ATP7A. Excess copper is released from brain cells into the cerebrospinal fluid (CSF) and taken up by choroidal epithelial cells that form the blood-CSF barrier. Copper is either stored for potential transport into CSF by ATP7B or is released into the bloodstream by ATP7A protein, thus a defect of ATP7B protein will cause accumulation of copper in the brain tissue [6].
CLINICAL PRESENTATION
Clinical features associated with symptomatic WD are highly variable and require a high index of suspicion for diagnosis. While the typical age of onset is from adolescence to early adulthood, neurologic symptoms tend to develop later by approximately one decade than hepatic presentation. Only 3.8% of patients have onset after 40 [7].
Initial signs and symptoms of WD are hepatic in approximately 40%, neurological in about 40-50% and psychiatric in about 10% [8]. Similar to our patient, many patients with neurologic symptoms do not have hepatic symptoms, thus the diagnosis of WD should not be ruled out based on age or absence of obvious liver pathology.
Hepatic symptoms in WD are similar to ones caused by other liver diseases, such as chronic liver disease, liver fibrosis, cirrhosis with portal hypertension or fulminant hepatic failure. The hepatic phenotype of WD is often associated with Coombs-negative hemolytic anemia.
Neurological manifestations usually mentioned in the literature are dystonic syndrome, pseudosclerotic (tremor, ataxia) syndrome, parkinsonian syndrome and dysarthric syndrome, although most patients have combinations. The most common initial neurological symptoms are dysarthria, dystonia, abnormal gait, tremor, parkinsonism, choreoathetosis and seizures [9,10].
There are many types of tremor in WD: hallmark classical “wing-beating” tremor (proximal tremor, appearing when the patient holds semi-flexed outstretched) and seen in our patient, rest tremor, postural and kinetic tremor, intention tremor and most prevalent non-rhythmic dystonic tremor [9,10].
Dystonia is frequently seen in WD and can be of any distribution and severity: focal dystonia (rarely isolated neck dystonia or writer’s cramp), multifocal, segmental or generalized. Generalized dystonia is characteristically seen in a combination with dysphagia, dysarthria or anarthria, severe gait disturbances and secondary skeletal changes in patients with a dystonic form of WD. Dysarthria is seen in almost all patients with WD and was also obvious on our patient’s first visit. It usually has a mixed dystonic and hypokinetic nature. It is typically accompanied by orofacial dyskinesias, lingual dystonia and so-called “risus sardonicus” – involuntary grimace with open mouth and contracted lips [9,10].
Parkinsonism is seen in up to half of patients and sometimes can have main Parkinson’s disease features like bradykinesia, tremor, hypomimia and shuffling gate [9,10]. Also, cerebellar ataxia, choreoathetosis and pyramidal signs can also be seen. Our patient presented with mainly parkinsonian features mixed with dystonia. The presence of sensory symptoms makes WD diagnosis unlikely. Epileptic seizures, usually generalized tonic-clonic with or without focal onset, can occur at any time during the course of WD, but more often after starting treatment and especially during neurological worsening that can sometimes be seen after initiating chelation treatment.
PSYCHIATRIC MANIFESTATIONS
Psychiatric problems can occur on treatment or in drug-naïve patients. The main symptoms are apathy, reduced attention, bradyphrenia, executive dysfunction, impaired social judgment and personality changes. Patients can occasionally manifest frank psychosis and be misdiagnosed as schizophrenia or bipolar disease. Cognitive problems can be present as global decline
KAYSER–FLEISCHER RINGS
Kayser–Fleischer rings represent a typical ophthalmological sign of WD that is seen in almost all patients with a neurological manifestation of WD [12]. They represent an asymptomatic copper deposition in Descemet’s corneal membrane and have brownish color that forms a full or partial circle. In light eyes they can be obviously visible like you see in the picture (Fig. 1A). Anyway, definitive detection of KFR is possible only using a slit lamp. The KFR can be absent or not fully formed in almost half of the patients with an isolated hepatic presentation. Like other symptoms of this disease, they disappear on treatment (Fig. 1B).
OTHER SYMPTOMS
Wilson’s disease affects multiple systems: blood (thrombocytopenia, hemolytic anemia and leucopenia, vitamin K deficiency), kidneys (nephrolithiasis, aminoaciduria), skin (hyperpigmentation of lower extremities, xerosis), endocrine system (delayed puberty, gynecomastia, glucose intolerance, parathyroid insufficiency), bones (osteoporosis, bone demineralization), heart (arrhythmias, cardiomyopathy) and eyes (KFR, sunflower cataract) [12].
DIAGNOSIS
Low ceruloplasmin is characteristic of WD, although this can rarely be a result of liver insufficiency of another cause. Occasionally, false low levels of ceruloplasmin can be seen due to oral contraceptive intake or inflammation. Other laboratory findings that support the diagnosis of WD are low plasma copper level, elevated transaminases, aminoaciduria, and hemolytic anemia. Important but simple and in many cases a diagnostic test is copper excretion in 24-hour urine. Excretion of > 100 μg/24h (1.6 μmol/24h) in the absence of cholestatic liver disease is characteristic for WD. Levels > 40 μg/24h (0.64 μmol/24h) are seen in asymptomatic children with WD [13]. Although validated only in children, a penicillamine test can be used to support a diagnosis: a 7-fold increase in copper excretion after two 500 mg doses of penicillamine [14].
Liver biopsy is occasionally used to diagnose a neurological form of WD if other tests are inconclusive. It shows copper levels of more than 250 μg/g of dry liver tissue (N=20-50 μg/g) [12]. A highly specific test of binding of ceruloplasmin with radioactive copper (64Cu) that shows weak binding in patients with WD unfortunately is usually unavailable. Family screening for WD is mandatory with parents, asymptomatic siblings and offspring being examined, ideally, also genetically.
Almost all patients with WD have some changes on brain MRI such as T2 hyperintensity of putamen, thalamus, midbrain, and pons. It is interesting to see a regress of MRI abnormalities on the treatment, in some cases a complete normalization.
Genetic testing gradually becomes more available and is used with laboratory tests for diagnosis. Due to so many mutations, a negative result of the genetic test does not rule out WD.
TREATMENT
Treatment for WD is life-long. It is impossible to control copper levels only with diet and its value is questionable, especially for the patient who is on WD treatment.
The mainstay of the therapy is chelating agents, which bind copper in the blood flow and tissues, providing its excretion, such as penicillamine that was successfully used in our patient, trientine and tetrathiomolybdate. Another option is zinc acetate that decreases intestinal absorption of copper and facilitates its removal by creating a negative balance. Choosing from these two options one should consider monitoring of some treatment-related risks. Although penicillamine is one of the most potent medications for WD, its use in some cases is associated with the initial worsening of neurological function in about 20% of patients [15]. Some of these patients never return to their previous condition. Most probably, this effect is a consequence of mobilization of a large amount of copper after initiation of the treatment and it is advised to start from low doses and increase slowly. In addition, there are treatment-resistant patients with irreversible impairment of the brain who never improve on the treatment. Although much safer, zinc acetate has lower potency, especially for hepatic forms. It can be comparable to penicillamine when used for a long time [16]. Zinc acetate is particularly appropriate for maintenance therapy after initial improvement is achieved and constant and also for life-long treatment of asymptomatic patients. A liver transplant is a method of choice in rare cases.
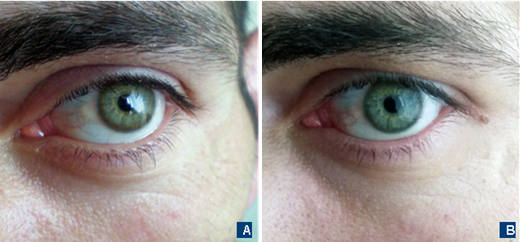
Fig. 1. The eyes of the patient with Wilson’s disease. A – Clearly visible Kayser–Fleischer rings before treatment. B – Washing out of Kayser–Fleischer rings on the treatment with penicillamine.
SUMMARY
We present a typical case of WD and its treatment. It is important to encourage general practitioners and especially neurologists to keep this disease in mind. Wilson’s disease is more prevalent than reviously thought and requires a high suspicion rate. It is easy to miss but it is also one of few potentially treatable diseases. Wilson’s disease should be ruled out in any person with unexplained hepatic, neurological or psychiatric disorders, especially under 40.
Conflict of interest: None.
Financial source: None.
REFERENCES
Olivarez L, Caggana M, Pass KA, Ferguson P, Brewer GJ. Estimate of the frequency of Wilson’s disease in the US Caucasian population: a mutation analysis approach. Ann Hum Genet. 2001;65(Pt 5):459-63.
Hahn SH, Lee SY, Jang YJ et al. Pilot study of mass screening for Wilson’s disease in Korea. Mol Genet Metab. 2002;76(2):133-6.
Lai CH, Tseng HF. Population-based epidemiologic study of Wilson’s disease in Taiwan. Eur J Neurol. 2010;17(6):830-3.
Ohura T, Abukawa D, Shiraishi H et al. Pilot study of screening for Wilson disease using dried blood spots obtained from children seen at outpatient clinics. J Inherit Metab Dis. 1999;22(1):74-80.
Coffey AJ, Durkie M, Hague S et al. A genetic study of Wilson’s disease in the United Kingdom. Brain. 2013;136(Pt 5):1476-87.
Scheiber IF, Brůha R, Dušek P. Pathogenesis of Wilson disease. Handb Clin Neurol. 2017;142:43-55.
Ferenci P, Członkowska A, Merle U et al. Late-onset Wilson’s disease. Gastroenterology. 2007;132(4):1294-8.
Pellecchia MT, Criscuolo C, Longo K et al. Clinical presentation and treatment of Wilson’s disease: a singlecentre experience. Eur Neurol. 2003;50(1):48-52.
Machado A, Chien HF, Deguti MM et al. Neurological manifestations in Wilson’s disease: Report of 119 cases. Mov Disord. 2006;21(12):2192-6.
Burke JF, Dayalu P, Nan B et al. Prognostic significance of neurologic examination findings in Wilson disease. Parkinsonism Relat Disord. 2011;17(7):551-6.
Svetel M, Pekmezović T, Petrović I et al. Long-term outcome in Serbian patients with Wilson disease. Eur J Neurol. 2009;16(7):852-7.
Członkowska A, Litwin T, Dusek P et al. Wilson disease. Nat Rev Dis Primers. 2018;4(1):21.
Nicastro E, Ranucci G, Vajro P, Vegnente A, Iorio R. Re-evaluation of the diagnostic criteria for Wilson disease in children with mild liver disease. Hepatology. 2010;52(6):1948-56.
Martins da Costa C, Baldwin D, Portmann B et al. Value of urinary copper excretion after penicillamine challenge in the diagnosis of Wilson’s disease. Hepatology. 1992;15(4):609-15.
Weiss KH, Thurik F, Gotthardt DN et al; EUROWILSON Consortium. Efficacy and safety of oral chelators in treatment of patients with Wilson disease. Clin Gastroenterol Hepatol. 2013;11(8):1028-35.e1-2.
Linn FH, Houwen RH, van Hattum J, van der Kleij S, van Erpecum KJ. Long-term exclusive zinc monotherapy in symptomatic Wilson disease: experience in 17 patients. Hepatology. 2009;50(5):1442-52.