The skin serves as a critical and effective barrier against a variety of harmful environmental stimuli, including mechanical, physical, chemical, thermal, bacterial, and immune factors [51,72]. Exposure to these damaging factors can lead to injury to viable tissues, disrupting their anatomical and physiological integrity [26,43,67]. In the context of skin, such damage typically involves the epithelial layer of the epidermis, resulting in compromised function of the underlying tissues and the formation of skin wounds [34,64]. To repair these injuries, the body initiates a complex, multistep process encompassing hemostasis, the inflammatory response, epithelial cell proliferation (including migration and differentiation), cellular interactions, the synthesis of extracellular matrix components, and the activation of various signaling pathways [34,72]. In chronic wounds, however, these regenerative processes are disrupted, and healing does not progress through normal stages. Instead, the process becomes stalled at the inflammatory phase, preventing the wound from achieving stable anatomical and functional resolution within the expected time frame, which is typically 3 months. As a result, these wounds persist and become resistant to conventional healing methods.
Wound healing can be delayed by multiple factors, including chronic diseases, vascular insufficiency, diabetes, malnutrition, aging, and local conditions such as pressure, infection, and edema [21,72]. Smoking is also a significant exogenous factor that impacts wound repair [36]. Given these influences, chronic wounds often have a secondary nature [72]. Definitions of chronic wounds vary, with some researchers considering any wound that persists beyond 6 weeks as chronic, whereas others define chronicity on the basis of wounds that fail to heal within 4-8 weeks according to normal regenerative sequences [21]. The reasons for impaired healing of certain wounds are multifaceted. Many chronic wounds stall in the inflammatory phase of healing. Factors contributing to the chronicity of wounds include underlying diseases, local hypoxia at the wound site, wound infection, and the presence of biofilms, and elevated levels of inflammatory mediators such as matrix metalloproteinases (MMPs) [3,21].
Currently, chronic wounds are classified into three main types on the basis of their underlying conditions: diabetic foot ulcers, venous ulcers of the lower limbs, and pressure ulcers [3,4,43]. These wound types share common characteristics, including stagnation in the inflammatory phase of healing and increased levels of MMPs. In addition, these wounds are frequently infected and often harbor bacterial biofilms [14,16]. Despite the diverse underlying metabolic disorders, effective treatment of chronic wounds largely depends on managing the underlying condition, which can potentially accelerate the healing process.
Chronic wounds pose a significant and growing challenge for patients, healthcare providers, and healthcare systems globally. In the United States, approximately 4.5 million individuals are estimated to be affected by chronic wounds [22], with 1.7% of those individuals aged 65 and older suffering from venous ulcers [1]. The burden of chronic, nonhealing wounds is substantial, impacting approximately 6.5 million patients annually in the United States and representing an escalating economic threat to public health, with annual treatment costs surpassing $9.7 billion [6]. The prevalence of wound-related diseases in the United Kingdom is approximately 2.2 million, which equates to 4.5% of the adult population [3]. Among these, venous ulcers of the legs are particularly notable, affecting 0.12% of individuals in the same age population and increasing to 1.2% in those aged 70 and older in Ireland [1].
The humanitarian and economic burden of chronic wounds is substantial and growing, driven by an aging population and the increasing prevalence of chronic diseases [41]. Chronic wounds are associated with significant pain, risk of infection, loss of function, and considerable financial costs. They frequently lead to severe outcomes such as amputations or sepsis. Despite their serious impact, chronic wounds are often overshadowed by their underlying causes. The costs associated with chronic wounds are inadequately documented, and there is a notable lack of comprehensive care and education on the subject. Chronic wounds thus remain a silent epidemic, significantly affecting the quality of life of more than 40 million people worldwide [72].
DISRUPTIONS IN CHRONIC WOUNDS AT THE TISSUE LEVEL
As previously mentioned, chronic wounds, due to the influence of various exogenous and endogenous factors, often fail to progress through the key stages of wound healing (hemostasis, inflammation, and repair) and frequently become stalled in the inflammatory phase [21, 72]. Under conditions of ongoing tissue damage, there is notable infiltration of neutrophils into the wound, leading to increased levels of reactive oxygen species (ROS) and the activity of destructive enzymes, which perpetuate the inflammatory cycle. However, addressing the primary harmful factor can effectively treat many chronic wounds, but its sustained and long-term impact may eventually lead to cellular senescence. Additionally, local tissue hypoxia, continuous damage, and heavy bacterial loads, combined with cellular and systemic responses to stress, perpetuate the cycle of damage, preventing progression to the proliferative phase of healing [72].
To elucidate the pathophysiological mechanisms underlying cellular disruptions in chronic wounds, it is crucial to first understand the tissue disturbances characteristic of these wounds. As previously outlined, the healing process in chronic wounds is often compromised by the interaction of several key factors. These include local tissue hypoxia, bacterial colonization and biofilm formation, and disruptions in tissue ischemia/reperfusion [21,72].
Chronic wounds frequently arise in the context of local tissue hypoxia, which is often associated with vascular abnormalities such as atherosclerosis, varicose veins, venous hypertension, or wound fibrosis, all of which contribute to reduced perfusion [63,72]. Impairments in wound blood flow lead to a relative decrease in oxygen perfusion pressure, resulting in hypoxic conditions within the wound. Chronic hypoxia plays a critical role in the development of nonhealing wounds and facilitates bacterial colonization within the wound cavity. The interplay between hypoxia and infection perpetuates a vicious cycle that is challenging to resolve [21].
The effects of tissue hypoxia disrupt cellular connections and activate inflammatory cascades. The increased expression of endothelial adhesion molecules, leads to neutrophil and macrophage extravasation. This process is accompanied by the autocrine production of proinflammatory cytokines, including IL-1α, IL-1β, IL-6, and TNF-α, which further exacerbate the inflammatory response [44,72].
The disruption of two critical balances is evident in chronic wounds: the balance between proteases and their inhibitors and the equilibrium between ROS and antioxidant systems. The overproduction of ROS leads to oxidative stress, resulting in lipid peroxidation and increased expression of specific factors, such as serine proteases, matrix metalloproteinases, and various inflammatory cytokines. Under conditions of local hypoxia, there is a reduction in the synthesis of nitric oxide (NO), which, despite its role as an antioxidant, is also involved in the activation of NF-κB [57,72]. Moreover, local hypoxia not only exacerbates inflammatory responses but also impairs key processes such as epithelialization and collagen synthesis in fibroblasts [55,72]. These disruptions further aggravate the problem and contribute to ongoing tissue degradation.
The etiopathogenesis of chronic wounds is significantly influenced by local ischemic changes, particularly those resulting from alterations in the ischemia/reperfusion relationship. In conjunction with tissue hypoxia, ischemia induces a proinflammatory state, as previously described. During reperfusion, excessive leukocytes, including neutrophils accumulate, migrate to the wound site and produce inflammatory cytokines. These ischemia-reperfusion cycles perpetuate, with their harmful effects intensifying, ultimately leading to tissue necrosis and ulceration [72].
Moreover, chronic wounds often present with a thickened, hyperproliferative epidermis that contains mitotically active cells. In contrast, the underlying tissue shows impaired proliferation of keratinocytes and other cellular colonies [30,59]. These tissue changes significantly impact cellular migration. Chronic wounds are characterized by the infiltration of mononuclear cells, including macrophages, lymphocytes, and plasma cells, as well as local macrophage proliferation at the inflammatory site. This is accompanied by the clonal proliferation of sensitized lymphocytes and their effector functions. Additionally, incomplete regeneration and fibrosis contribute to structural disruptions within the wound [72].
In addition to the aforementioned factors, wound infection is a significant issue in chronic wounds. Wound colonization by microorganisms is characterized by the presence of proliferating organisms within the wound without causing immediate damage to the host tissue. This colonization begins at the initial stages of wound formation and subsequently contributes to tissue damage, leading to wound infection [21]. In chronic wounds, microorganisms often form biofilms [3], which are discussed in detail further in this article.
MICROBIOME OF CHRONIC WOUNDS
In chronic wounds, in addition to tissue and cellular disruption, the third critical pathogenic factor is mandatory colonization by bacteria [3,21,72]. Successful wound closure has been shown to depend on the presence of fewer than 105 organisms per gram of tissue; however, chronic wounds often harbor bacterial counts exceeding 105 organisms [21,72]. Factors that increase the likelihood of infection include immune system suppression, malnutrition, hypoxia (whether due to arterial or venous insufficiency), and the presence of foreign bodies and necrotic tissue, which act as reservoirs for infection.
Local hypoxia is a primary contributor to bacterial colonization in chronic wounds. Numerous studies have documented an inverse relationship between infection rates and wound oxygenation levels. This phenomenon is likely attributable to the oxygen-dependent activity of antimicrobial enzymes within neutrophils, such as myeloperoxidase. Consequently, periods of ischemia in chronic wounds impair the bactericidal mechanisms of the host [72].
The etiology of chronic wounds can be diverse, but infections caused by various microbial species directly impair the normal healing process, leading to wound persistence [62]. Common pathogens associated with wound infections include Staphylococcus aureus, Pseudomonas aeruginosa, and β-hemolytic streptococci, all of which are known to delay wound healing and contribute to chronic wound infections [27].
Empirical data from numerous independent culture studies indicate that Gram-positive cocci (GPC) are the most frequently identified microorganisms in chronic wounds among diabetic foot patients. Among these strains, Staphylococcus aureus is consistently the most prevalent and is found in more than 50% of wounds, followed by coagulase-negative Staphylococci spp. [27,33]. S. aureus, including methicillin-resistant S. aureus (MRSA), is often found in conjunction with other Gram-positive pathogens and mixed anaerobic environments. In contrast to common misconceptions, Pseudomonas aeruginosa is not as prevalent as previously thought [24,31].
Certain fungi, such as Trichosporon asahii, have been implicated in the development of wound infections and have been identified in the exudate of chronic wounds. Frequently, fungi from the Candida and Cladosporium genera are found in wound exudates. The presence of fungal microflora can interact with the bacterial microflora, facilitating bacterial proliferation and dissemination. Furthermore, this interaction can lead to the formation of distinctive wound biofilms [23].
In addition to directly damaging the host, bacteria contribute to the recruitment of leukocytes. This process enhances the effects of inflammatory cytokines, proteases, and ROS, thereby both initiating and perpetuating inflammatory cascades [55].
BACTERIAL BIOFILMS IN CHRONIC WOUNDS
Although recent studies on the microbiota of chronic wounds have focused primarily on planktonic organisms (microorganisms that are free-floating or suspended in a liquid), these microorganisms can also be components of bacterial biofilms [9]. Research over the past decade has indicated that 99.9% of these microorganisms are capable of adhering to wound surfaces because of the nutrients present on those surfaces. Once on these surfaces, microorganisms begin to produce complex exopolymers containing polysaccharides, various proteins, and nucleic acids. These factors contribute to both the adhesion and colonization of microorganisms, as well as the establishment of close interactions between them [12].
Bacteria that colonize chronic wounds frequently form polymicrobial biofilms, where the synergy with accompanying microorganisms creates an optimal environment for bacteria. This environment allows bacteria to evade the host immune response and the effects of antibiotics [19,20].
Biofilms represent structurally complex, dynamic systems that offer a protected environment conducive to bacterial growth, thereby enabling microbial cells to survive and proliferate within chronic wounds. This process significantly contributes to the chronicity of the underlying pathological conditions [21]. Established biofilms meticulously modulate the host’s inflammatory response, extending its duration by providing a stable nutrient source derived from the inflammatory exudate. Concurrently, biofilms act as persistent reservoirs of pathogen-associated molecular patterns, thereby perpetuating the inflammatory response [9].
A biofilm is characterized by a form of syntrophic cooperation among microorganisms [19], manifesting as a three-dimensional mosaic consortium of bacteria [18]. In this consortium, cells adhere to one another through their surfaces. These adherent cells are encased in a thin extracellular matrix composed of extracellular polymeric substances [19]. The biofilm-associated cells produce these polymeric substances, which include extracellular polysaccharides, proteins, lipids, and DNA polymeric conglomerates [19,29]. As a three-dimensional structure and a community of microorganisms, biofilms are metaphorically referred to as “microbial cities” [66].
Microorganisms form biofilms due to certain environmental factors, which include specific and nonspecific receptors on cell surfaces, nutrient factors, and, in some cases, antibiotic concentrations [20]. The hydrophobicity of bacteria can also significantly impact their ability to establish biofilms. Bacteria exhibiting high hydrophobicity tend to have reduced repulsion towards surfaces, facilitating adhesion [12]. Owing to their limited motility, certain bacterial species may be incapable of directly adhering to surfaces; however, they can anchor to the existing matrix or other bacterial colonies. Compared with their motile counterparts, non-motile bacteria generally exhibit less efficiency in surface recognition and attachment [12].
During the colonization of surfaces, bacterial cells communicate through quorum-sensing signals, such as N-acylhomoserine lactones. Upon the initiation of colonization, biofilm development proceeds through processes of cell division and accumulation [29,42]. Studies have particularly investigated quorum-sensing signals in biofilms associated with chronic wounds, focusing on microorganisms such as Staphylococcus aureus and Pseudomonas aeruginosa. In these instances, quorum sensing is mediated by autoinductive peptides [42].
The process of biofilm formation induces changes in the bacterial phenotype, resulting in alterations in gene regulatory mechanisms [2].
The involvement of bacterial biofilms in chronic wound pathogenesis is now well documented. However, notable variability exists in the bacterial colonization of chronic wounds. Pathogenic bacteria frequently emerge as the dominant microflora, supplanting more universally present species [42]. Oxygen limitations, which extend deeper into the biofilm, facilitate the proliferation of anaerobic species within chronic wounds [61]. Consequently, focusing exclusively on the bacterial load within the wound is insufficient; it is crucial to also evaluate the species present and their interactions within the wound milieu. This includes determining whether these species are competing for growth or survival.
Research highlights recurring patterns of combined species that demonstrate synergistic interactions, leading to chronic wound infections. These interactions are referred to as “functional equivalent pathogen groups,” and the presence of such groups in wounds can number in the hundreds [28].
Recent research underscores the significant role that fungi play in the development and persistence of biofilms within chronic wounds. Notably, in chronic wounds such as those associated with diabetic foot ulcers, where the normal skin microbiota includes fungi, biofilms often result from the interactions between bacteria and fungi. Candida albicans, a prevalent member of the foot microbiota, has been shown to form complex microbiota in conjunction with bacterial species including Staphylococcus aureus, Pseudomonas aeruginosa, Burkholderia cenocepacia, Streptococcus spp., Acinetobacter baumannii, Enterococcus faecalis, and Escherichia coli.
In these settings, bacteria can proliferate on the fungal hyphal surfaces, leading to the formation of stable biofilms. Additionally, these fungal hyphae may invade adjacent epithelial cells persistently, facilitating bacterial penetration into these cells. The bacteria also cover themselves with fungal polysaccharides, which enhances their adhesion, aggregation, and tolerance [23]. This mechanism contributes to the chronicity of infections by fostering bacterial resistance to antibiotics. Furthermore, bacterial enzymatic activity degrades critical components such as fibrin and growth factors essential for wound healing [21].
Notably, bacterial biofilms can remain macroscopically invisible, as demonstrated by biofilms formed by Trichosporon asahii and Staphylococcus simulans in diabetic foot ulcers, which ultimately leads to limb amputation [23].
Several hypotheses have been proposed to explain the persistence of bacterial biofilms against therapeutic agents. One hypothesis suggests that the primary factor is the inherent heterogeneity of the biofilm structure [35]. Alternatively, the presence of cell-persisters within the biofilm may enable its resuscitation posttreatment [28]. Additionally, the stability of biofilms may be attributed to the inherent resistance of microorganisms to antibiotics and the presence of multidrug-resistant bacterial strains [32].
DISRUPTIONS IN CHRONIC WOUNDS AT THE CELLULAR LEVEL
Given the diverse primary causes of chronic wound formation outlined above, examining cellular disruptions on a case-by-case basis is pertinent.
Venous Ulcers
Chronic venous disease is underpinned by complex etiological and pathophysiological processes. The condition has a multifactorial etiology, encompassing genetic predisposition, environmental factors, and alterations in venous endothelial function, inflammatory mediators, and structural changes in the vascular wall. These factors collectively contribute to the development of dilated, tortuous veins, valvular insufficiency, venous hypertension, and their subsequent clinical manifestations, notably chronic venous ulcers [49].
The impact on microcirculation commences with the effects of altered shear stress on the endothelium. This leads to an increased production of vasoactive substances by endothelial cells, upregulation of selectins, and increased synthesis of inflammatory molecules, chemokines, and prothrombotic mediators [54]. Bergan et al. (2006) have identified that mechanical forces, including low shear stress and stretch, result in an increased synthesis of intercellular adhesion molecule-1 (ICAM-1, CD54), vascular cell adhesion molecule-1 (VCAM-1, CD-106), endothelial leukocyte adhesion molecule-1 (CD62, E-selectin), and mechanosensitive vanilloid transient potential receptors by endothelial cells. This heightened expression is particularly evident under conditions of venous hypertension associated with chronic venous insufficiency [5]. These adhesion molecules mediate the inflammatory process and facilitate the migration of leukocytes to the vein wall and valves [17, 56]. Consequently, an inflammatory cascade is activated, characterized by increased cytokine production and matrix metalloproteinase (MMP) expression. MMPs target fibroblasts, vascular smooth muscle cells, and the extracellular matrix [45]. Elevated expression of TNF-α, IL-1α, IL-6, TGF-β1, PDGF-A, EGF, βFGF, and VEGF has been documented in keratinocytes adjacent to venous ulcers [48]. Additionally, the levels of MMP-1, MMP-2, MMP-3, MMP-9, and MMP-13 are notably elevated in both tissue and plasma samples [69] (Figure 1).
These processes result in the proliferation of smooth muscle cells, which lose their contractility and collagen-synthesizing ability. This proliferation leads to hypertrophic segments with reduced contractility, increased rigidity, and impaired elasticity. Thus, the response of the vein wall and its ability to maintain physiological function under elevated venous pressure are compromised [47]. Studies have demonstrated that in varicose veins, there is a predominance of type I collagen synthesis versus type III collagen synthesis, which accounts for changes in the extensibility of varicose veins [53].
Hypertrophic segments with modified smooth muscle cells and increased extracellular matrix content are observed alongside atrophic regions in the vein wall, which are characterized by lower extracellular matrix and smooth muscle cell content [38]. Additionally, altered collagen synthesis and reduced cellular proliferation due to abnormal responses to TGF-β1 signaling and aging impact fibroblasts [10] (Figure 1).
Furthermore, Crawford et al. (2017) highlighted additional factors contributing to decreased contractility, including changes in endothelial cell endothelin B receptors, reduced levels of cyclic adenosine monophosphate, and discrepancies in the levels of prostacyclin and thromboxane A2 [10].
Venous hypertension and low shear stress on the endothelial surface can instigate a pathological cascade that results in adverse changes in the venous wall, venous valves, and surrounding skin, ultimately leading to venous dilation and the development of venous ulcers.
Diabetic Foot Ulcers
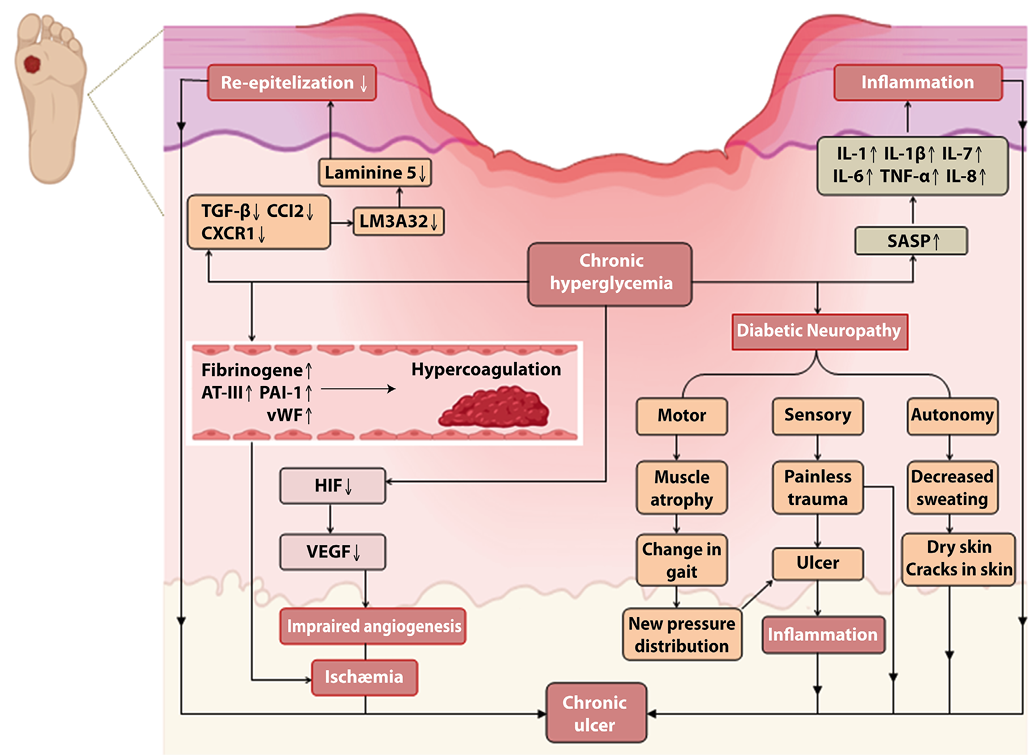
The hyperglycemic environment in diabetes mellitus significantly impacts various stages of wound healing. Evidence indicates that diabetic patients exhibit elevated levels of fibrinogen, antithrombin III (AT-III), plasminogen activator inhibitor-1 (PAI-1), and von Willebrand factor activity, placing them in a state of hypercoagulability and hypofibrinolysis [15]. Thrombogenesis and occlusion of damaged blood vessels during the wound healing process create hypoxic conditions, which in turn stimulate the production of hypoxia-inducible factor 1-alpha (HIF-1α). This factor, in conjunction with HIF-1β, forms the HIF complex in the nucleus, thereby promoting the synthesis of various molecular factors, including vascular endothelial growth factor (VEGF), which is crucial for angiogenesis. However, in the context of diabetic foot ulcers, elevated glucose levels can increase local osmotic pressure, potentially diminishing HIF-1α production and reducing the blood supply [7] (Figure 2).
In addition to inflammatory states and impaired angiogenesis, diabetes disrupts progenitor cell recruitment, proliferation, and the release of growth factors postinjury from a cellular standpoint [50]. Neutrophils, which typically act as first-line defense cells, display reduced functional activity under chronic hyperglycemic and hyperinsulinemic conditions, increasing susceptibility to infections and exacerbating their severity [58].
The hyperglycemic milieu also accelerates cellular aging and promotes the senescence-associated secretory phenotype (SASP) in macrophages, which contributes to the secretion of proinflammatory cytokines (e.g., IL-1α, IL-6, IL-7, and IL-8), growth factors (e.g., TGF-β), and proteases (e.g., matrix metalloproteinases), thus altering the tissue microenvironment [68] (Figure 2). Macrophage-specific cytokines, such as MCP-1, are integral components of SASP, and SASP factors influence classical macrophage receptors (e.g., CXCR1, CXCR2, CX3CR1) [52]. Furthermore, the increased production of proinflammatory cytokines may be linked to elevated levels of NF-κB IL1B, IL6, and IL8 in hyperinsulinemic states, which augment the expression of inflammation-associated genes [58].
In diabetic patients, cytokine production extends beyond inflammatory cells to include skin keratinocytes and microvascular endothelial cells. Research on diabetic foot ulcers has demonstrated elevated levels of TGF-β1 and its receptor TGFβR1 in keratinocytes, as well as increased levels of CCL2, CXCR1, and TGFβR1 in skin endothelial cells [46]. Keratinocyte alterations also involve decreased production of LM-3A32, a precursor to laminin isoforms, including laminin-5, which is essential for epithelial cell adhesion to the basal membrane and their motility through integrins, which are crucial for re-epithelialization [7].
Failure to transition from the inflammatory to the proliferative phase may result from activation of the p38 mitogen-activated protein kinase signaling pathway, leading to cytokine production and a reduction in miR-21 levels, which is critical for resolving the inflammatory phase [8].
In chronic wounds, the accumulation of apoptotic cells due to increased advanced glycation end-products (AGEs), protein kinase C activation, and oxidative stress—exacerbated by impaired macrophage phagocytic activity—intensifies the inflammatory response [25]. Oxidative stress is particularly influential in the development of peripheral neuropathy. Studies have validated patterns of lipid peroxidation product accumulation, depletion of glutathione (GSH), and reduced superoxide dismutase activity in peripheral nerves, alongside the identification of novel markers of damage from AGEs, such as decreased catalase activity and increased superoxide and nitrotyrosine production [40]. Antioxidant treatments have proven effective in addressing the changes in these markers associated with diabetes [40].
Pressure Ulcers
Pressure ulcers, also known as decubitus ulcers, commonly arise when sustained pressure on the skin and underlying tissues occurs between a bony prominence and an external surface, such as a mattress or wheelchair cushions. The etiology of pressure ulcers involves several factors, including the ischemic-reperfusion injury mechanism (Figure 3). This mechanism posits that external pressures exceeding the arterial perfusion pressure (approximately 32 mmHg) and the venous outflow pressure (approximately 8-12 mmHg) impair blood circulation, leading to local tissue hypoxia [37]. During ischemic periods, reduced nitric oxide (NO) production induces vasoconstriction, whereas during reperfusion, increased NO production results in vasodilation. However, extensive damage during reperfusion can cause the release of toxic metabolites and reactive oxygen species, further injuring surrounding tissues and leading to endothelial cell damage. Consequently, this process diminishes the initial increase in NO production by endothelial cells during reperfusion [60]. Blood reperfusion, which leads to cellular edema, tissue damage, and excessive production of reactive oxygen species, triggers oxidative stress. Oxidative stress can result in the accumulation of unfolded proteins in the endoplasmic reticulum (ER) lumen, thereby disrupting ER homeostasis and causing ER stress [11]. Research by Wang et al. has demonstrated that mitochondrial-mediated apoptosis may play a role in the early stages of pressure ulcer formation, with HIF-1α contributing to its activation. Elevated levels of matrix metalloproteinase-9 (MMP-9) have also been observed in experimental models of ischemic-reperfusion injury [65].
Lymphatic vessels are crucial for the removal of toxic metabolites and excess fluid from the interstitial space. The obstruction of these vessels in the context of pressure ulcers contributes to inflammation and cell death. Lymphedema, a component of the pathophysiology of pressure ulcers, is associated with elevated levels of proinflammatory molecules, including TNF-α, IL-6, IL-8, and monocyte chemoattractant protein-1 (MCP-1) (Figure 3). This exacerbates the inflammatory response by promoting the infiltration of proinflammatory cells and impeding the resolution of inflammation due to the decreased activity of T-regulatory cells [70].
Recent investigations into pressure ulcers have identified disrupted inflammatory responses linked to the microRNAs miR-21 and miR-885-3p. Initial suppression of miR-21 in a lipopolysaccharide-induced model of pressure ulcer keratinocytes leads to increased expression of proinflammatory markers. Conversely, treatment with emodin upregulates miR-21, inhibits NFκB signaling, and reduces the levels of IL-6, IL-1β, COX-2, and iNOS. This effect is attributed to enhanced macrophage efferocytosis, a transition to an anti-inflammatory phenotype, activation of PI3K/AKT signaling, and improved keratinocyte viability [39]. Additionally, overproduction of miR-885-3p contributes to reduced NFκB activity and suppression of TLR-4, thereby mitigating the inflammatory response [71] (Figure 3).
conclusion
The pathophysiology of chronic wounds reveals both distinct and overlapping factors that affect wound healing, underscoring the complexity of their management. Each type of chronic wound has unique etiological and cellular dynamics; however, notable commonalities exist that require further investigation to refine our understanding of the underlying processes and to identify targeted therapeutic strategies.
Key features of chronic wounds include diminished angiogenesis, impaired epithelialization, and excessive production of ROS. An analysis of these processes across the cellular and subcellular levels revealed that all chronic wounds are characterized by persistent inflammation and manifestations of all three phases of the wound healing continuum. The wound bed typically displays a combination of fibrin and granulation tissue, with potential areas of necrosis and purulent discharge. Granulation tissue is often described as pale and mottled, with wound edges and surrounding tissues exhibiting increased firmness, whereas marginal epithelialization is infrequently observed.
Alterations in cellular and extracellular matrix interactions are evident in chronic wounds. The chronic inflammatory infiltrate, which predominantly consists of monocyte-macrophage cells, along with elevated levels of plasma cells and T and B lymphocytes, disrupts the normal wound healing process. Additionally, an imbalance in the ratio of T-helper to T-regulatory cells is observed. Chronic nonhealing wounds show reduced expression of type I and III procollagen mRNA in dermal fibroblasts due to phenotypic changes. This fibroblast “senescence” leads to decreased proliferative activity and diminished synthesis of extracellular matrix components, with evidence suggesting that procollagen synthesis inhibition correlates with collagen fiber accumulation in the dermis.
In addition, decreased expression of PDGF and its receptors, increased TNF-α expression, and reduced levels of TGF-β and its receptors contribute to delayed wound healing. These changes lead to diminished extracellular matrix component synthesis and impaired fibroblast-to-myofibroblast conversion, resulting in compromised wound contraction.
Dysregulation of cytokines and growth factors is another critical factor that is influenced by their excessive utilization relative to normal or elevated synthesis levels. This imbalance disrupts the intracellular enzymatic systems responsible for extracellular matrix remodeling, leading to increased fibronectin levels, altered proteoglycan ratios, and reduced interstitial collagen content.
Recent advancements in treatment modalities reflect these insights, with a significant focus on collagen-based materials that act as matrices for tissue regeneration. Upon application, collagen preparations interact with the wound, fibroblasts, blood and lymphatic vessels, and embedded nerve fibers, thereby facilitating matrix alignment and promoting wound healing.
Addressing underlying conditions and enhancing patient quality of life are essential components of effective chronic wound management. For example, in cases of diabetic foot, optimizing glucose levels and continuous monitoring are critical. Despite these advances, chronic wound treatment remains a major challenge in clinical practice, significantly impacting the quality of life of millions of patients globally. Further research is needed to refine treatment strategies and improve patient outcomes in the management of chronic wounds.
REFERENCES
- Abelyan G, Abrahamyan L, Yenokyan G. A case-control study of risk factors of chronic venous ulceration in patients with varicose veins. Phlebology. 2018;33(1):60-67
- An D, Parsek M. The promise and peril of transcriptional profiling in biofilm communities. Curr Opin Microbiol. 2007;10(3):292-6
- Atkin L. Chronic wounds: the challenges of appropriate management. Br J Community Nurs. 2019;24(S9):S26-S32
- Barrigah-Benissan K, Ory J, Sotto A et al. Antiseptic agents for chronic wounds: a systematic review. Antibiotics (Basel). 2022;11(3):350
- Bergan JJ, Schmid-Schönbein GW, Smith PD et al. Chronic venous disease. N Engl J Med. 2006;355(5):488-498
- Bickers DR, Lim HW, Margolis D et al. The burden of skin diseases: 2004: A joint project of the American Academy of Dermatology Association and the Society for Investigative Dermatology. J Am Acad Dermatol. 2006;55(3):490-500
- Blakytny R, Jude EB. Altered molecular mechanisms of diabetic foot ulcers. Int J Low Extrem Wounds. 2009;8(2):95-104
- Burgess JL, Wyant WA, Abdo Abujamra B et al. Diabetic wound-healing science. Medicina (Kaunas). 2021;57(10):1072
- Costerton JW, Cheng KJ, Geesey GG et al. Bacterial biofilms in nature and disease. Annu Rev Microbiol. 1987;41:435-464
- Crawford J, Lal B, Durán W, Pappas P. Pathophysiology of venous ulceration. J Vasc Surg Venous Lymphat Disord. 2017;5(4):596-605
- Cui FF, Pan YY, Xie HH et al. Pressure combined with ischemia/reperfusion injury induces deep tissue injury via endoplasmic reticulum stress in a rat pressure ulcer model. Int J Mol Sci. 2016;17(3):284
- Donlan RM. Biofilms: microbial life on surfaces. Emerg Infect Dis. 2002;8(9):881-890
- Eberhardt RT, Raffetto JD. Chronic venous insufficiency. Circulation. 2014;130(4):333-346
- Edmonds M, Lázaro-Martínez JL, Alfayate-García JM et al. Sucrose octasulfate dressing versus control dressing in patients with neuroischaemic diabetic foot ulcers (Explorer): an international, multicentre, double-blind, randomised, controlled trial. Lancet Diabetes Endocrinol. 2018;6(3):186-196
- Erem C, Hacihasanoğlu A, Celik S et al. Coagulation and fibrinolysis parameters in type 2 diabetic patients with and without diabetic vascular complications. Med Princ Pract. 2005;14(1):22-30
- Frykberg RG, Banks J. Challenges in the treatment of chronic wounds. Adv Wound Care (New Rochelle). 2015;4(9):560-582
- Fujita H. Molecular biology of adhesion molecules – structure, expression and function of ICAM-1 and ELAM-1. Nihon Rinsho. 1993;51(6):1643-1649
- Gilbert P, Maira-Litran T, McBain AJ et al. The physiology and collective recalcitrance of microbial biofilm communities. Adv Microb Physiol. 2002;46:202-256
- Hall-Stoodley L, Costerton JW, Stoodley P. Bacterial biofilms: from the natural environment to infectious diseases. Nat Rev Microbiol. 2004;2(2):95-108
- Hoffman LR, D’Argenio DA, MacCoss MJ, Zhang Z, Jones RA, Miller SI. Aminoglycoside antibiotics induce bacterial biofilm formation. Nature. 2005;436(7054):1171-1175
- Izadi K, Ganchi P. Chronic wounds. Clin Plast Surg. 2005;32(2):209-222
- Jones RE, Foster DS, Longaker MT. Management of chronic wounds-2018. JAMA. 2018;320(14):1481-1482
- Kalan L, Grice EA. Fungi in the Wound Microbiome. Adv Wound Care (New Rochelle). 2018;7(7):247-255
- Kalan LR, Brennan MB. The role of the microbiome in nonhealing diabetic wounds. Ann N Y Acad Sci. 2019;1435(1):79-92
- Khanna S, Biswas S, Shang Y et al. Macrophage dysfunction impairs resolution of inflammation in the wounds of diabetic mice. PLoS One. 2010;5(3):e9539
- Kujath P, Michelsen A. Wounds - from physiology to wound dressing. Dtsch Arztebl Int. 2008;105(13):239-248
- Lebowitz D, Gariani K, Kressmann B et al. Are antibiotic-resistant pathogens more common in subsequent episodes of diabetic foot infection? Int J Infect Dis. 2017;59:61-64
- Lewis K. Persister cells and the riddle of biofilm survival. Biochemistry (Mosc). 2005;70(2):267-274
- Li S, Liu SY, Chan SY, Chua SL. Biofilm matrix cloaks bacterial quorum sensing chemoattractants from predator detection. ISME J. 2022;16(5):1388-1396
- Lindley L, Stojadinovic O, Pastar I, Tomic-Canic M. Biology and biomarkers for wound healing. Plast Reconstr Surg. 2016;138:18S-28S
- Loesche M, Gardner SE, Kalan L et al. Temporal Stability in Chronic Wound Microbiota Is Associated With Poor Healing. J Invest Dermatol. 2017;137(1):237-244
- Mah T, Pitts B, Pellock B et al. A genetic basis for Pseudomonas aeruginosa biofilm antibiotic resistance. Nature. 2003;426:306-10
- Malone M, Bowling FL, Gannass A et al. Deep wound cultures correlate well with bone biopsy culture in diabetic foot osteomyelitis. Diabetes Metab Res Rev. 2013;29(7):546-550
- Masson-Meyers D, Andrade T, Caetano G et al. Experimental models and methods for cutaneous wound healing assessment. Int J Exp Pathol. 2020 Feb;101(1-2):21-37
- McBain AJ, Allison D, Gilbert P. Emerging strategies for the chemical treatment of microbial biofilms. Biotechnol Genet Eng Rev. 2000;17:267-279
- McDaniel JC, Browning KK. Smoking, chronic wound healing, and implications for evidence-based practice. J Wound Ostomy Continence Nurs. 2014;41(5):415-E2
- Mervis JS, Phillips TJ. Pressure ulcers: Pathophysiology, epidemiology, risk factors, and presentation. J Am Acad Dermatol. 2019;81(4):881-890
- Metcalfe MJ, Baker DM, Turmaine M, Burnstock G. Alterations in purinoceptor expression in human long saphenous vein during varicose disease. Eur J Vasc Endovasc Surg. 2007;33(2):239-250
- Niemiec SM, Louiselle AE, Liechty KW, Zgheib C. Role of microRNAs in pressure ulcer immune response, pathogenesis, and treatment. Int J Mol Sci. 2020;22(1):64
- Obrosova IG. Update on the pathogenesis of diabetic neuropathy. Curr Diab Rep. 2003;3(6):439-445
- Olsson M, Järbrink K, Divakar U et al. The humanistic and economic burden of chronic wounds: A systematic review. Wound Repair Regen. 2019;27(1):114-125
- Omar A, Wright JB, Schultz G, Burrell R, Nadworny P. Microbial Biofilms and Chronic Wounds. Microorganisms. 2017;5(1):9
- Pang M, Zhu M, Lei X et al. Microbiome imbalances: an overlooked potential mechanism in chronic nonhealing wounds. Int J Low Extrem Wounds. 2019;18(1):31-41
- Peschen M, Lahaye T, Hennig B et al. Expression of the adhesion molecules ICAM-1, VCAM-1, LFA-1 and VLA-4 in the skin is modulated in progressing stages of chronic venous insufficiency. Acta Derm Venereol. 1999;79(1):27-32
- Pocock ES, Alsaigh T, Mazor R, Schmid-Schönbein GW. Cellular and molecular basis of venous insufficiency. Vasc Cell. 2014;6(1):24
- Pradhan L, Nabzdyk C, Andersen N et al. Inflammation and neuropeptides: the connection in diabetic wound healing. Expert Rev Mol Med. 2009;11:e2
- Raffetto JD, Ligi D, Maniscalco R et al. why venous leg ulcers have difficulty healing: overview on pathophysiology, clinical consequences, and treatment. J Clin Med. 2020;10(1):29
- Raffetto JD. Inflammation in chronic venous ulcers. Phlebology. 2013;28(S1):61-67
- Raffetto JD. Pathophysiology of Chronic Venous Disease and Venous Ulcers. Surg Clin North Am. 2018;98(2):337-347
- Rodrigues M, Kosaric N, Bonham CA, Gurtner GC. Wound Healing: A Cellular Perspective. Physiol Rev. 2019;99(1):665-706
- Rousselle P, Montmasson M, Garnier C. Extracellular matrix contribution to skin wound re-epithelialization. Matrix Biol. 2019;75-76:12-26
- Sagiv A, Krizhanovsky V. Immunosurveillance of senescent cells: the bright side of the senescence program. Biogerontology. 2013;14(6):617-628
- Sansilvestri-Morel P, Rupin A, Badier-Commander C et al. Chronic venous insufficiency: dysregulation of collagen synthesis. Angiology. 2003;54(S1):S13-S18
- Schmid-Schönbein GW, Takase S, Bergan JJ. New advances in the understanding of the pathophysiology of chronic venous insufficiency. Angiology. 2001;52(S1):S27-S34
- Schreml S, Szeimies RM, Prantl L et al. Oxygen in acute and chronic wound healing. Br J Dermatol. 2010;163(2):257-268
- Singh V, Kaur R, Kumari P et al. ICAM-1 and VCAM-1: Gatekeepers in various inflammatory and cardiovascular disorders. Clin Chim Acta. 2023;548:117487
- Soneja A, Drews M, Malinski T. Role of nitric oxide, nitroxidative and oxidative stress in wound healing. Pharmacol Rep. 2005;57(S):108-119
- Stegenga ME, van der Crabben SN, Dessing MC et al. Effect of acute hyperglycaemia and/or hyperinsulinaemia on proinflammatory gene expression, cytokine production and neutrophil function in humans. Diabet Med. 2008;25(2):157-164
- Stojadinovic O, Brem H, Vouthounis C et al. Molecular pathogenesis of chronic wounds: the role of beta-catenin and c-myc in the inhibition of epithelialization and wound healing. Am J Pathol. 2005;167(1):59-69
- Struck BD, Wright JE. Pressure ulcers and endothelial dysfunction: is there a link? J Nutr Elder. 2007;26(3-4):105-117
- Thomson CH. Biofilms: do they affect wound healing? Int Wound J. 2011;8(1):63-67
- Tipton CD, Sanford NE, Everett JA et al. Chronic wound microbiome colonization on mouse model following cryogenic preservation. PLoS One. 2019;14(8):e0221565
- Toledo-Pereyra LH, Lopez-Neblina F, Toledo AH. Reactive oxygen species and molecular biology of ischemia/reperfusion. Ann Transplant. 2004;9(1):81-83
- Wang M, Huang X, Zheng H et al. Nanomaterials applied in wound healing: Mechanisms, limitations and perspectives. J Control Release. 2021;337:236-247
- Wang Y, Pu L, Li Z, Hu X, Jiang L. Hypoxia-Inducible Factor-1α Gene Expression and Apoptosis in Ischemia-Reperfusion Injury: A Rat Model of Early-Stage Pressure Ulcer. Nurs Res. 2016;65(1):35-46
- Watnick P, Kolter R. Biofilm, city of microbes. J Bacteriol. 2000;182(10):2675-2679
- Wilkins RG, Unverdorben M. Wound cleaning and wound healing: a concise review. Adv Skin Wound Care. 2013;26(4):160-163
- Wilkinson HN, Clowes C, Banyard KL, Matteuci P, Mace KA, Hardman MJ. Elevated Local Senescence in Diabetic Wound Healing Is Linked to Pathological Repair via CXCR2. J Invest Dermatol. 2019;139(5):1171-1181.e6
- Xiao Y, Huang Z, Yin H, Lin Y, Wang S. In vitro differences between smooth muscle cells derived from varicose veins and normal veins. J Vasc Surg. 2009;50(5):1149-1154
- Yoshida S, Koshima I, Hamada Y et al. Lymphovenous anastomosis aids wound healing in lymphedema: relationship between lymphedema and delayed wound healing from a view of immune mechanisms. Adv Wound Care (New Rochelle). 2019;8(6):263-9
- Zhang X, Gu H, Wang L et al. MiR-885-3p is down-regulated in peripheral blood mononuclear cells from T1D patients and regulates the inflammatory response via targeting TLR4/NF-κB signaling. J Gene Med. 2020;22(1):e3145
- Zhao R, Liang H, Clarke E et al. Inflammation in Chronic Wounds. Int J Mol Sci. 2016;17(12):2085

Figure 1. Pathophysiologic mechanism of venous ulcers. Venous hypertension and low shear stress on the endothelial surface can instigate a pathological cascade that results in adverse changes in the venous wall, venous valves, and surrounding skin, ultimately leading to venous dilation and the development of venous ulcers.
Pathophysiology of chronic wounds
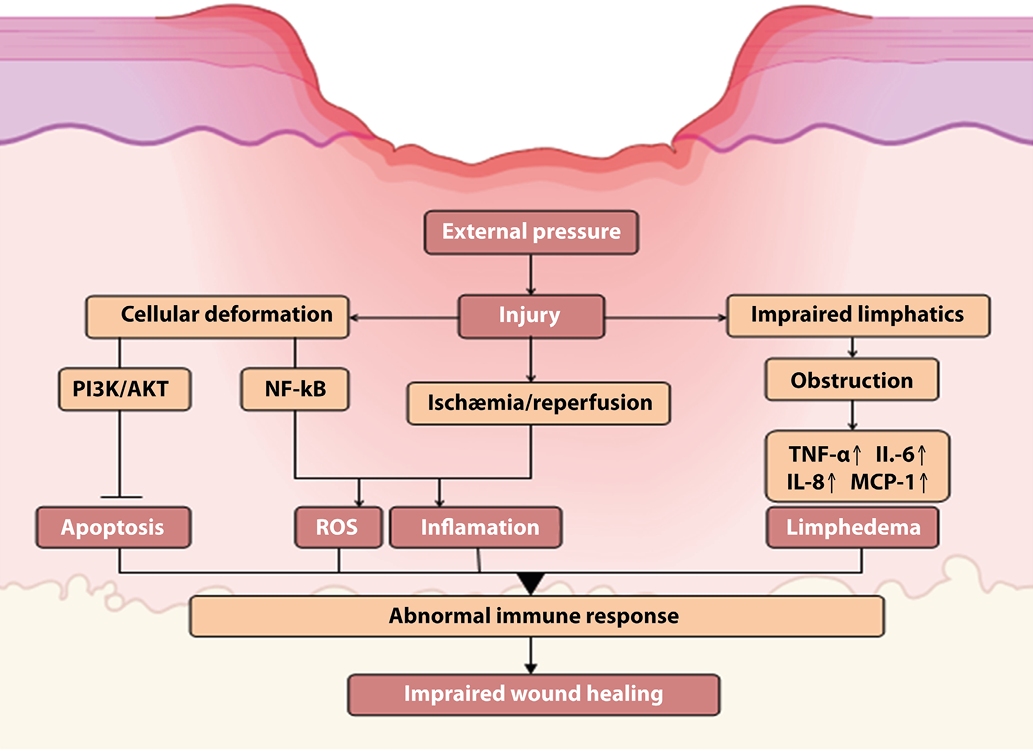

Figure 2. Pathophysiologic mechanism of diabetic foot ulcer. The pathophysiological processes underlying diabetic foot ulcers involve impaired angiogenesis, neuropathy, disrupted inflammatory responses, and barrier function compromise, contributing to the formidable challenges associated with chronic wound healing.
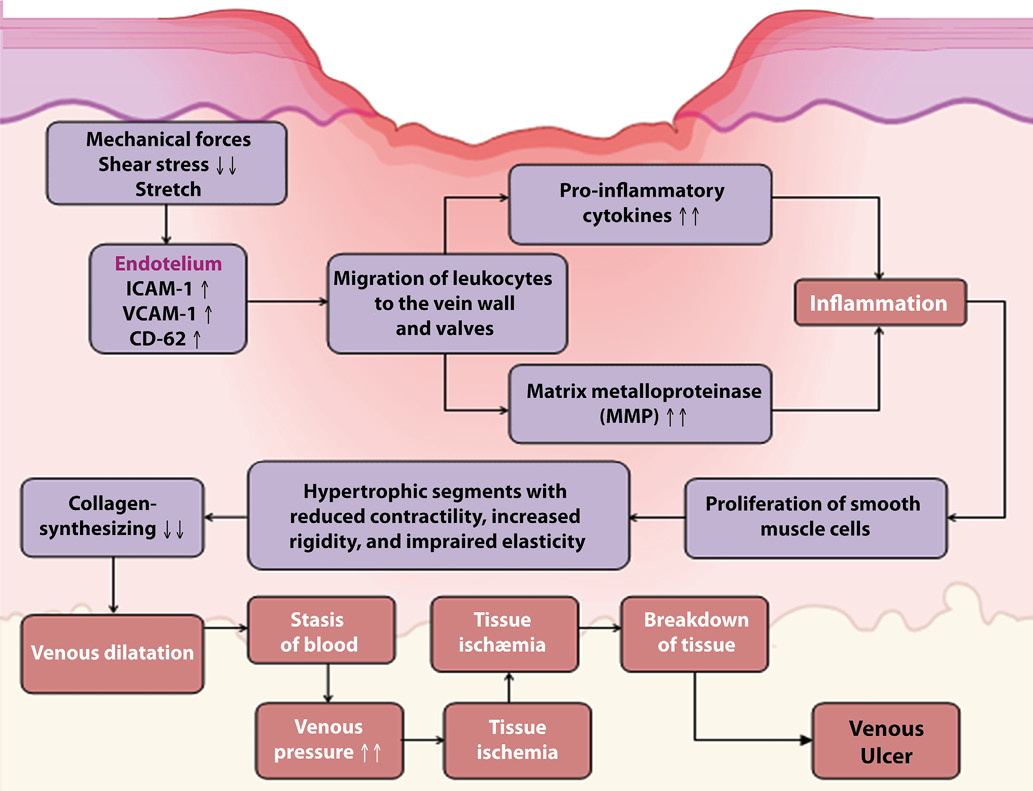

Figure 3. Pathophysiologic mechanism of pressure ulcers. Ischemia-reperfusion, lymphatic channel obstruction, and cellular deformation result in elevated inflammation, ROS, and apoptosis, which contribute to immune response dysregulation and impaired wound healing. Flat head arrows indicate inhibition, and pointed arrows indicate activation.